Module RemoteBlast
Date de publication : 22 janvier 2009 , Date de mise à jour : 04 Mars 2009
Par
Jasmine Minguet
Cet article présente le module RemoteBlast qui permet l'exécution à
distance de Blast (Basic Local Alignment Search Tool) d'NCBI via HTTP.
Blast est un algorithme de comparaison de séquences utilisé pour rechercher
dans des base de données de séquences les alignements locaux optimaux
à une séquence entrée par l'utilisateur.
I. Fonctionnement du Blast
II. Code :
III. Liens utiles :
IV. Références utilisées afin de rédiger cet article :
I. Fonctionnement du Blast
Dans certains cas, l'alignement global ne révèle pas la similarité attendue. Par exemple, les séquences suivantes :
- G G C T G A C C A C C T T
- G A T C A C T T C C A T G
produiraient l'alignement global suivant :

alors que le résultat attendu aurait plutôt ressemblé à cela :
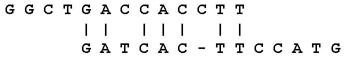
Le programme BLAST permet de faire un alignement global. Il est utilisé pour rechercher une séquence similaire à une séquence requête dans une banque de données de séquences. Le programme commence par déterminer tous les mots de taille w (w valant 3 pour les protéines et 11 pour les acides nucléiques) présents dans la séquence requête. On parle de hachage. Pour chaque mot, une liste de mots voisins est générée. Chaque mot voisin de la liste possède un score supérieur ou égal à un seuil T (généralement égal à 13).
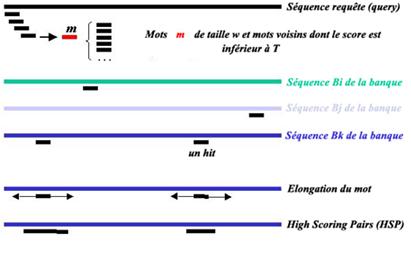
Chaque mot de la liste de mots est ensuite comparé à tous les mots
de taille w de chaque séquence de la banque de données. Lorsqu'un
mot d'une séquence de la banque est identique à un mot de la liste
de mot voisins, un hit est enregistré. Pour chaque hit, le programme
effectue une extension sans gap de l'alignement dans les deux sens.
L'extension s'arrête quand le score du mot étendu diminue de plus
qu'un seuil X fixé. Les segments ayant un score de similarité supérieur
à un score S seuil fixé sont retenus (High Scoring Pairs = HSP).
Exemple
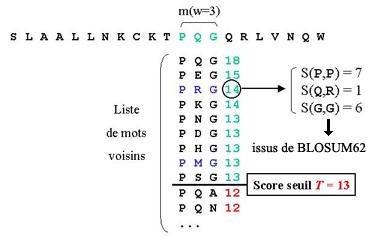
Soit la séquence suivante : S L A A L L N K C K T P Q G Q R L V N Q W
Le but est de trouver des séquences similaires à cette séquence dans une banque de données de séquences comme SwissProt par exemple. Après le hachage de la séquence avec une taille de mot w = 3 et une constitution de la liste des mots voisins, le programme recherche dans les séquences de la banques des mots identiques. Nous nous sommes limité à une explication sur le mot PQG :
Lorsqu'un hit est enregistré, le mot est allongé de part et d'autre tant que le score ne diminue pas de plus d'un seuil X fixé :
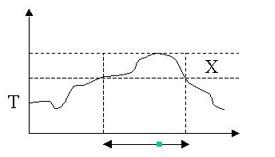
Lorsque le seuil est atteint, l'élongation s'arrête et on obtient la paire de segments de haut score ou High Scoring Pairs (HSP) :

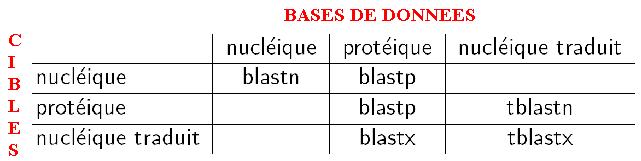
Plusieurs algorithmes sont disponibles :
Définitions importantes :
Expected value = E-value : le nombre de différents alignements avec un score équivalent ou meilleur que S étant attendu par hasard dans la base de données. Au plus la E-value est petite au plus le score est significatif.
P-value : probabilité qu'un alignement ait un score égal ou supérieur au score en question. La P-value est calculée en mettant en relation le score observé de l'alignement (S) avec la distribution des scores HSP provenant de comparaisons de séquences générées aléatoirement, de même longueur et composition que la séquence cible. Les P-values les plus significatives sont celles proches de zéro. P-value et E-value sont deux façons différentes de représenter la signifiance de l'alignement.
Expected value et P-value sont dépendant de plusieurs facteurs, incluant le système de score employé, la composition de la séquence cible, une composition en résidus pour les séquences de la base de données supposée similaire à celle de la séquence cible et de la longueur totale de la base de données.
II. Code :
Lorsque l'on utilise ce module, la première chose à faire est de le charger.
use strict;
use Bio::Tools::Run::RemoteBlast;
|
Différents algoritmes sont disponibles :
blastn compare une séquence cible nucléique à une base de données nucléique.
blastp compare une séquence cible protéique à une base de données protéique.
cf tableau dans l'introduction
Blast query tutorial
Base de données dont les entrées seront comparées à notre séquence cible
dans ce cas-ci nr (base par défaut) contenant les séquences non redondantes
pour la liste complète des bases de données, se rendre sur
Blast tutorial query
Expected value (défaut 10)
Méthode utilisée afin de lire le rapport du blast
my $readmethod = 'SearchIO';
|
Passage des paramètres au module
my @params = (
'-prog' = $prog,
'-data' = $db,
'-expect' = $e_val,
'-readmethod' = $readmethod );
my $factory = Bio::Tools::Run::RemoteBlast->new(@params);
|
Séquence cible entrée sous le format Bio::Seq<br/>
my $sequence = 'GCCTCACGGTCCTGCTGATATGTGACATCACCCCCGGAGGCCCAGCTGTAAATTCCTCTCTTTGTACTCTTTC';
my $input = Bio::Seq->new( -display_id => 'identifiant', -seq => $sequence);
my $r = $factory->submit_blast($input);
|
... ou passage d'un fichier contenant une ou plusieurs séquences à analyser.<br/>
my $r = $factory-;submit_blast('chemin/fichier.fa');
|
Option d'affichage : $v permet d'activer ou de désactiver les messages
my $v = 1;
print STDERR "waiting..." if( $v > 0 );
while ( my @rids = $factory->each_rid ) {
foreach my $rid ( @rids ) {
my $rc = $factory->retrieve_blast($rid);
if( !ref($rc) ) {
if( $rc < 0) {
$factory->remove_rid($rid);
}
print STDERR "." if ( $v > 0 );
sleep 5;
}
else {
my $result = $rc->next_result();
my $filename = 'chemin'.$result->query_name().'.blastn';
$factory->save_output($filename);
$factory->remove_rid($rid);
while ( my $hit = $result->next_hit ) {
next unless ( $v > 0);
print "\thit name is ", $hit->name, "\n";
while( my $hsp = $hit->next_hsp ) {
print "\t\tscore is ", $hsp->score, "\n";
}
}
}
}
}
|
Exemple du premier hit trouvé dans variable $result
'_newhits_below_threshold' => [
{
'-algorithm' => 'BLASTN',
'-score' => '132',
'-description' => 'Homo sapiens bile acid Coenzyme A: amino acid '
.'N-acyltransferase (glycine N-choloyltransferase) (BAAT), transcript variant 1, mRNA',
'-length' => '3478',
'-query_len' => '73',
'-hsp_factory' => bless( {
'interface' => 'Bio::Search::HSP::HSPI',
'type' => 'Bio::Search::HSP::GenericHSP',
'_loaded_types' => {
'Bio::Search::HSP::GenericHSP' => 1
},
'_root_verbose' => 0
}, 'Bio::Factory::ObjectFactory' ),
'-rank' => 1,
'-name' => 'ref|NM_001701.3|',
'-hsps' => [
{
'-query_start' => '1',
'-algorithm' => 'BLASTN',
'-hit_seq' => 'GCCTCACGGTCCTGCTGATATGTGACATCACCCCCGGAGGCCCAGCTGTAAATTCCTCTCTTTGTACTCTTTC',
'-hit_length' => '3478',
'-query_length' => '73',
'-query_desc' => '',
'-query_frame' => 0,
'-rank' => 1,
'-hit_desc' => 'Homo sapiens bile acid Coenzyme A: amino '
.'acid N-acyltransferase (glycine N-choloyltransferase) (BAAT), transcript variant 1, mRNA',
'-hsp_gaps' => '0',
'-query_end' => '73',
'-hit_name' => 'ref|NM_001701.3|',
'-identical' => '73',
'-query_name' => 'identifiant',
'-evalue' => '1e-28',
'-score' => '146',
'-conserved' => '73',
'-hit_frame' => 0,
'-hsp_length' => '73',
'-query_seq' => 'GCCTCACGGTCCTGCTGATATGTGACATCACCCCCGGAGGCCCAGCTGTAAATTCCTCTCTTTGTACTCTTTC',
'-hit_start' => '1',
'-homology_seq' => '|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||',
'-hit_end' => '73',
'-bits' => '132'
}
],
'-accession' => 'NM_001701',
'-significance' => '1e-28'
},
|
Affichage dans le fichier de sortie
>ref|NM_001701.3| Homo sapiens bile acid Coenzyme A: amino acid N-acyltransferase
(glycine N-choloyltransferase) (BAAT), transcript variant
1, mRNA
Length=3478
Score = 132 bits (146), Expect = 1e-28
Identities = 73/73 (100%), Gaps = 0/73 (0%)
Strand=Plus/Plus
Query 1 GCCTCACGGTCCTGCTGATATGTGACATCACCCCCGGAGGCCCAGCTGTAAATTCCTCTC 60
||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||
Sbjct 1 GCCTCACGGTCCTGCTGATATGTGACATCACCCCCGGAGGCCCAGCTGTAAATTCCTCTC 60
Query 61 TTTGTACTCTTTC 73
|||||||||||||
Sbjct 61 TTTGTACTCTTTC 73
|
III. Liens utiles :
IV. Références utilisées afin de rédiger cet article :


Les sources présentées sur cette page sont libres de droits
et vous pouvez les utiliser à votre convenance. Par contre, la page de présentation
constitue une œuvre intellectuelle protégée par les droits d'auteur. Copyright ©
2009 Jasmine80. Aucune reproduction, même partielle, ne peut être
faite de ce site ni de l'ensemble de son contenu : textes, documents, images, etc.
sans l'autorisation expresse de l'auteur. Sinon vous encourez selon la loi jusqu'à
trois ans de prison et jusqu'à 300 000 € de dommages et intérêts.